自動メソッド開発のソフトウェアメーカー
![]()
![]() |
|![]()








ホーム > 公開講座 > HPLC基礎講座
HPLC基礎講座
2015年12月3日
クロムソードジャパン株式会社

参考:Agilent 1290 InfinityⅡ UHPLC
目次
- 1.クロマトグラフィーの原理
- 2.HPLCの装置構成
- 3.HPLCの分離モード
- 4.検出器
- 5.Q&A形式でご質問にお答えします。
- Q1.カラムの性能をチェックする方法は?
- Q2.一度調製した移動相を用いて、異なる濃度の移動相を再調製したいのですが?
- Q3.逆相分配クロマトグラフィーを行う際、カラムは何を基準に選べばよいですか?(準備中)
- Q4.各メーカーから様々な種類の逆相分配カラムがでていますが、その中から自分にあったカラムを選ぶには?
(準備中) - Q5.逆相分配カラムで解離しやすい官能基を持つ試料を分離する際の注意点は?
- Q6.イオンペアクロマトグラフィーについて教えてください。
- Q7.イオンペア試薬の使い方について教えてください。
- Q8.グラジエント分析法で、使用するカラムの長さは分離効率に影響しますか?
- Q9.グラジエント分析法で、使用するカラムの内径は分離効率に影響しますか?
- Q10.グラジエント分析法で、流速は分離効率に影響しますか?
- Q11.カラム使用後の洗浄、保存方法を教えてください。(準備中)
- Q12.HPLCに使用する有機溶媒の取り扱い注意点は?(準備中)
- Q13.移動相の脱気は必要ですか?
- Q14.移動相に緩衝液を使用する際の条件と注意点は?(準備中)
- Q15.よいカラムとはどんなカラムですか?(準備中)
- Q16.生体試料中に夾雑している高分子物質(たんぱく質、多糖類など)を除く方法は?(準備中)
- Q17.分離ピークのテーリング、リーディング、ブロード化の原因は?
- Q18.高極性物質をHPLCにより分析したいのですが、どのような方法がありますか?
- Q19.逆相カラムには多くの種類が市販されていますが、違いを教えてください。
- Q20.HPLCでアミノ酸を分析したいのですが、どのような方法がありますか?
- Q21.HPLCで陰イオン界面活性剤を分析したいのですが、どのような方法がありますか?(準備中)
- Q22.HPLCで非イオン界面活性剤を分析したいのですが、どのような方法がありますか?(準備中)
- Q23.HPLCで雨水中の無機イオンを分析したいのですが、どのような方法がありますか?
- Q24.HPLCで糖類を分析したいのですが、どのような方法がありますか?
- Q25.逆相HPLCの溶離液によく使われるメタノールとアセトニトリルの用途の違いは何ですか?
- Q26.カラムを長持ちさせる方法は?(準備中)
- Q27.カラムの圧力が上昇してしまいました。その原因と対処法は?(準備中)
- Q28.逆相HPLCで使用する溶離液中の有機溶媒の割合と保持時間との関係は?(準備中)
- Q29.順相HPLCで使用する溶離液中の有機溶媒の割合と保持時間との関係は?(準備中)
- Q30.分析開始前、溶離液をどの程度流せばカラムが安定しますか?(準備中)
- Q31.カラムを初めて使用する場合の取り扱い方、カラム使用後の洗浄、保存方法を教えてください。
- ※掲載準備中のQ&Aにつきましては、準備ができ次第、順次公開致します。
1.クロマトグラフィーの原理
(1)クロマトグラフィーとは
20世紀の初頭、ロシアの植物学者のTswettは、炭酸カルシウムの粉末を詰めたガラスカラムを用いて、植物抽出液を石油エーテルと共に流すと、図1のように色素が分離してくることを発見しました。彼はこの方法を、ギリシャ語の「色」と「描く」を意味する「chroma」「graphos」から「chromatographie」と命名し、混合試料中の各成分を分離、分析する方法として、現在広く普及しているクロマトグラフィーの礎を築きました。
(図1)
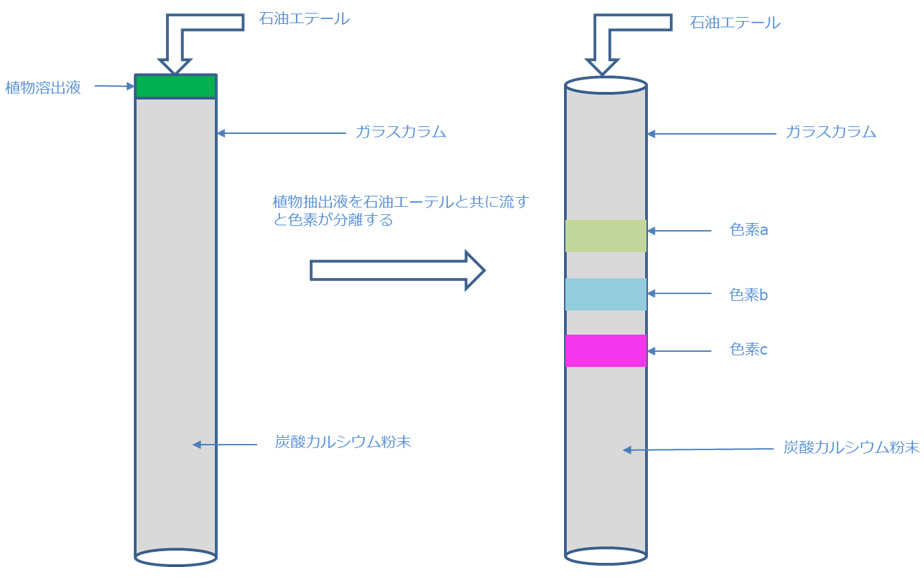
(2)専門用語
それぞれよく似た言葉ですが、
「クロマトグラフィー」:分離の方法。
「クロマトグラフ」:分離に用いる装置。
「クロマトグラム」:分離の結果として得られる、検出シグナルの強度を時間的経過とともにあらわした図。
例えば、「アジレントテクノロジー社製のクロマトグラフを用いて、食品中の色素添加物の高速液体クロマトグラフィーを行った結果、図に示すようなクロマトグラムが得られた。」の様に表現します。
(図:クロマトグラム例)
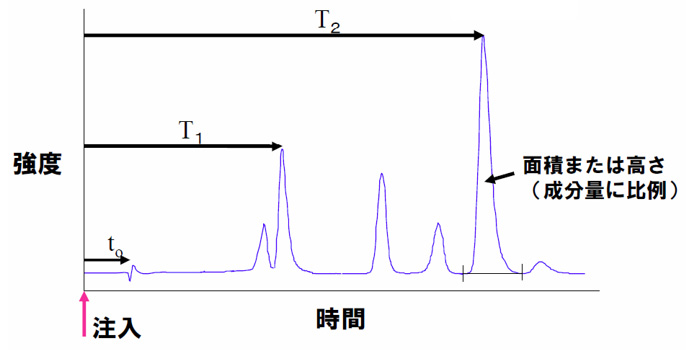
試料を分離するために、「固定相」、「移動相」と呼ばれる媒体が必要です。
「固定相」:固定された媒体、一般的にはカラム中の充填剤。
「移動相」:試料とともにカラムの固定相の中を移動する媒体、液体クロマトグラフィーでは、溶離液と呼びます。
(3)液体クロマトグラフィー(LC)とガスクロマトグラフィー(GC)
移動相が液体か、気体かによって、表のように2つに分類されます。

2.HPLCの装置構成
(1)HPLCの装置例(図および画像)
【HPLC本体】

【オートサンプラ(マルチサンプラ)】

【オートサンプラ例(マルチサンプラ)】
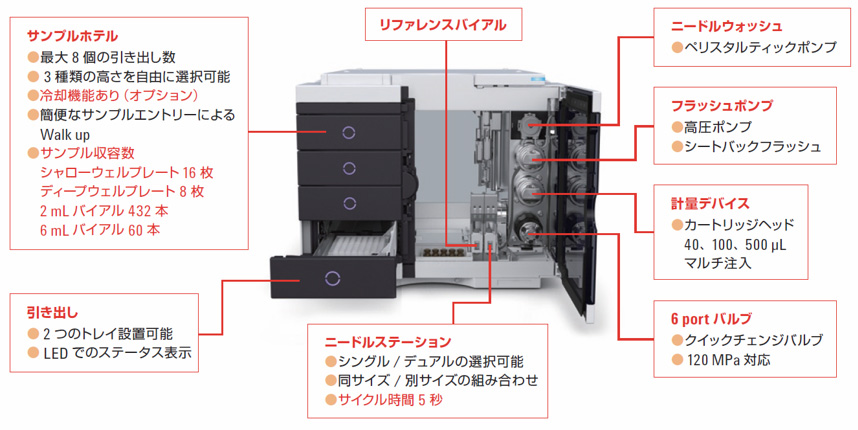
【カラムフィッティング】

【LCカラム】

【ダイオードアレイ検出器】

(2)送液部(ポンプ)
溶離液を一定流量で脈流(圧力変動)なく送液することが重要です。多成分分析の場合、一種類の溶離液で全ての成分をうまく分離するのは難しいので、分析途中で溶離液組成を変えながら分析を行う方法が考えられています。一定時間ごとに階段状に組成を切り替えるステップワイズ法と、時間とともに徐々に組成を変えていくグラジエント法があります。また、組成を変えないで分析する方法はアイソクラティック(イソクラ)法とよびます。
(3)試料注入部
試料を流路に注入する部分です。試料の注入にはマイクロシリンジを用い、インジェクターにマイクロシリンジを手動でセットして試料を注入するマニュアルインジェクターと、自動で注入するオートサンプラがあります。
(4)分離部
固定相カラムを用い、試料成分の分離目的に合わせてカラムを選択します。カラムの温度が変わると試料成分の分離や溶出時間に影響が生じますので、再現性の良いデータを得るためにはカラムオーブンを用い、常に一定温度で分析を行います。
(5)検出部
カラムから溶出した成分を検出して電気シグナルに変換する部分です。目的とする試料成分に応じて、様々な検出器が開発されています。主な検出器としては、吸光度検出器(UV検出器、UV-VIS検出器、フォトダイオードアレイ(PDA)検出器)、示差屈折率(RI)検出器、電気伝導度(CD)検出器、蛍光(FL)検出器などがあります。また、検出部に質量分析計(MS)を用いて物質の質量を測定することで物質を直接同定する方法も利用が広がっています。
(6)データ処理部
検出部で検出されたピークの高さあるいは面積を電気シグナルとしてレポートします。
3.HPLCの分離モード
試料が装置に注入されてカラムに入ると、まず充填剤(固定相)に保持されます。保持される度合いは成分により異なり、固定相に親和性の小さい成分ほど溶離液(移動相)と共に早くカラムの中を流れていきます。一方、固定相に親和性の大きい成分は長時間固定相に保持されるので、カラムの中をゆっくり流れていき移動が遅れます。このように各成分が固定相と移動相のどちらに親和性が大きいかによりカラムの中を移動する速さに差が生じ、その結果、各成分が分離されて出てくるのです。分離モードは主に吸着、分配、イオン交換、サイズ排除の4つに分類されますが、実際にはこれらのモードが複合的に作用して分離されると考えられています。
(1)吸着
固定相の充填剤としてシリカゲルやアルミナを用い、充填剤の表面に静電気的引力により物質が吸着することで分離が行われます。しかしこの充填剤にいったん吸着すると溶出してこない物質も多く、長く使用していると分離の再現性が悪くなるという欠点があります。
(2)分配
現在最もよく用いられている分離モードです。固定相と移動相間における試料成分の親和性(溶解性)の差により分離してきます。したがって、移動相より固定相に溶解しやすい成分ほど遅く溶出することになります。分配モードでは、固定相と移動相の極性の相対的な関係から順相系と逆相系に分けられます。
順相系:固定相の極性が移動相の極性より高い関係にあるものを順相系といいます。そのため、試料成分としては親水性(極性)化合物に適しています。したがって、移動相には、非水溶性有機溶媒または水溶性有機溶媒と水溶液(水、緩衝液)の混合溶液を使用します。固定相には、シリカゲルや合成ポリマーゲルを充填剤基材として、極性基で表面を修飾したものが多く用いられています。
逆相系:分配モードの中で最も広く用いられています。固定相の極性が移動相の極性より低い関係にあるものを逆相系といいます。試料成分としては中~低極性のほとんどの化合物に適用できます。したがって、移動相には、水溶性有機溶媒と水溶液(水、緩衝液)の混合溶液を使用し、有機溶媒の割合が増すほど溶出力は高くなります。固定相には、シリカゲルや合成ポリマーゲルを充填剤基材として、表面に炭素数が18(ODS)や8の疎水性のアルキル基が化学結合したものが多く用いられています。
【分配クロマトグラフィー】
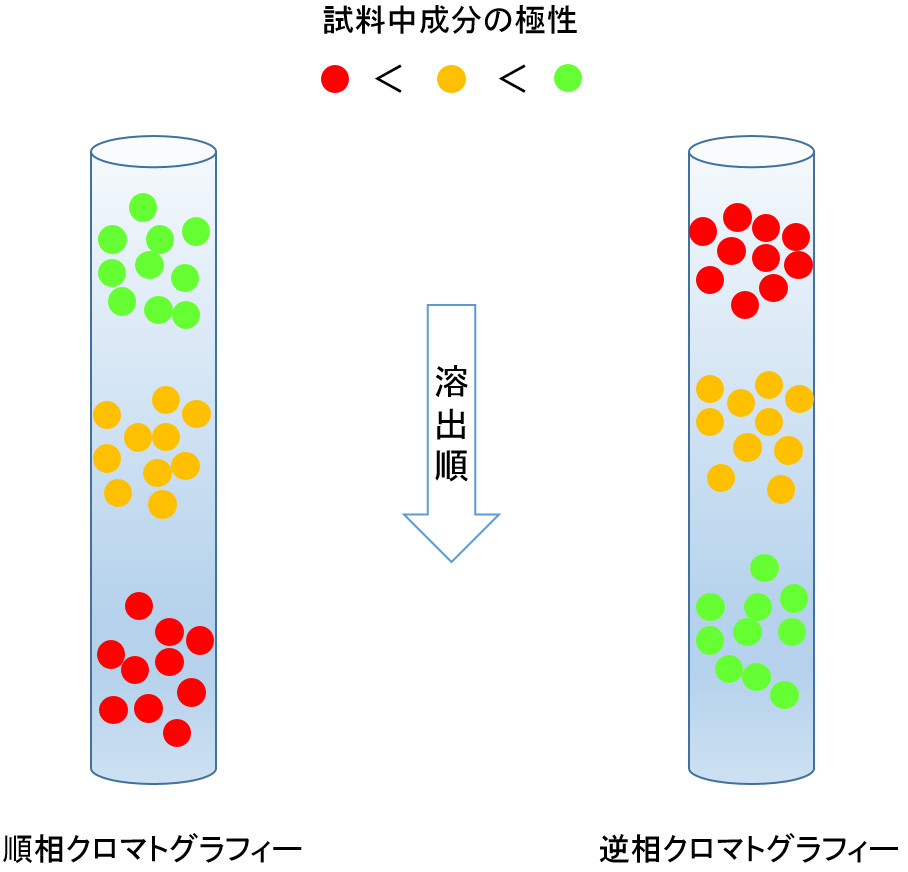
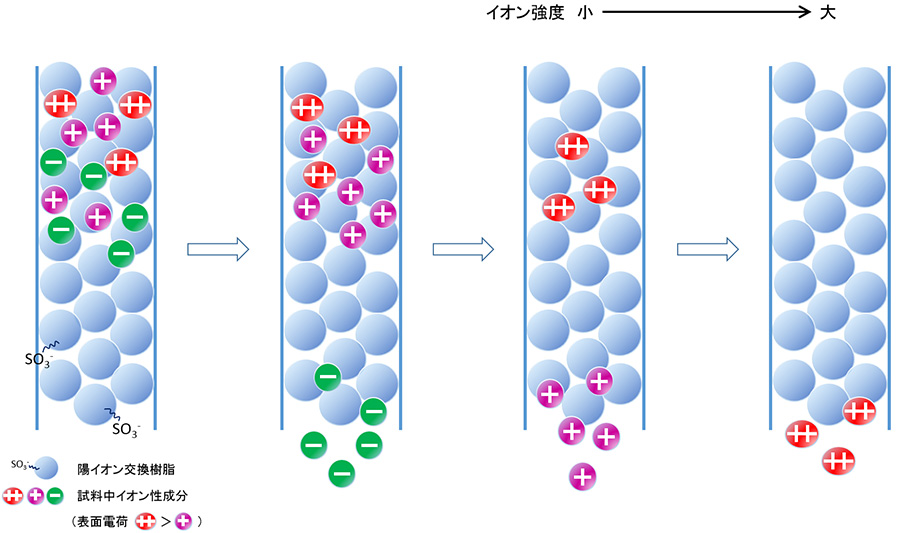
(3)イオン交換
物質の電荷の違いを利用する分離モードです。タンパク質や核酸のような水に溶けてイオンになる物質や無機イオンは、陰イオンについては陰イオン交換樹脂で、陽イオンは陽イオン交換樹脂で分離します。タンパク質を構成するアミノ酸は、陽イオンになるアミノ基(-NH2)と陰イオンになるカルボキシル基(-COOH)を持っていますので、分子の電荷が0になるpH(等電点)を挟んで、酸性側ではプラスに、塩基性側ではマイナスに帯電します。したがって、アミノ酸やタンパク質を分離する場合、溶離液のpHを変えることで、陽イオン交換、陰イオン交換を使い分けることができます。また近年、特に環境中の無機イオンの測定のために開発された方法はイオンクロマトグラフィーと呼ばれ、環境水の水質管理や大気環境測定などに広く適用されています。
【イオン交換クロマトグラフィー】
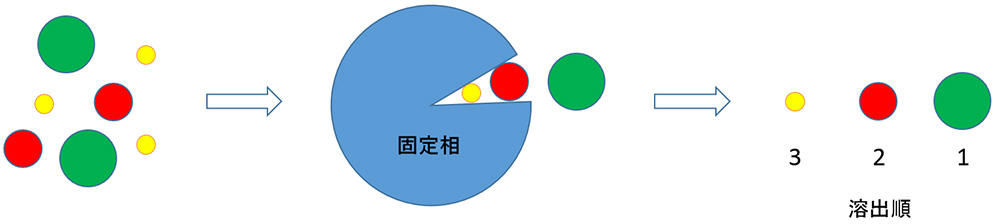
(4)サイズ排除
分子の大きさに基づいて分離する方法です。固定相には、架橋有機性高分子ゲルや多孔性シリカ粒子が用いられ、これらのゲルは粒子内に細孔を持ち、細孔より大きな分子は内部に浸透できないので、粒子間を素通りして早く溶出し、細孔より小さな分子は細孔に保持されて遅く溶出してきます。細孔のサイズの違いで、細孔内に浸透する分子でも溶出時間に違いが出てきます。ゲル浸透、ゲルろ過とも呼ばれています。
【サイズ排除クロマトグラフィー】
4.検出器
(1)UV/UV-VIS検出器
現在最も広く使用されている検出器です。紫外・可視領域(190nm~900nm)に吸光を持つ多くの有機物質が分析できます。UV検出器は、光源に重水素ランプ(D2ランプ)を用い190nm~380nmの吸光度を測定します。さらに長波長側(~900nm)の吸光度を測定する場合は、光源にタングステンランプ(Wランプ)を追加したUV-VIS検出器を用います。
(2)フォトダイオードアレイ(PDA)検出器
検出原理は、基本的にはUV-VIS検出器と同じです。しかし、UV-VIS検出器から得られるデータは時間軸と吸光度の二次元ですが、フォトダイオードアレイ検出器では、受光部に多数のフォトダイオードアレイを並べることで、一度に多波長のスペクトル情報を得ることができ、クロマトグラムは時間軸、吸光度、スペクトルの三次元データとなります。したがって、最適な波長を測定を繰り返すことなく、一度で求めることが可能となります。
(3)示差屈折率(RI)検出器
物質のもつ屈折率の違いを検出する測定です。ほとんどの化合物が溶媒とは異なる屈折率をもつため、あらゆる成分が検出可能ですが、感度が低いことが欠点です。また、温度変化や溶離液組成の変化によっても屈折率は変化するため、グラジエント分析ができません。
(4)電気伝導度(CD)検出器
溶液の電気伝導度を測定することで、溶液中のイオンを検出する方法です。一定電圧を印加した電極間をイオンが通る際に変化する電流値を測定します。イオンクロマトグラフィーにおける専用検出器として普及しています。高感度分析が可能ですが、温度変化の影響を受けやすいという欠点があります。
(5)蛍光(FL)検出器
物質が光を吸収してエネルギー準位の高い励起状態になった後、エネルギー準位の低い安定した基底状態に戻る際に発する蛍光を検出する方法です。もともと蛍光を持つ物質は多くありませんが、蛍光はその物質に特有ですので、蛍光検出は特異性が高く一般的に高感度です。
(6)LC-MS
液体クロマトグラフィー(LC)と質量分析法(MS)とを融合させた分析法です。質量分析で得られるマススペクトルから、分子の質量や構造、さらに同位体イオンピークの高さから構成元素の種類と数などの元素情報が得られ、物質の同定が可能になります。
5.Q&A形式でご質問にお答えします。
Q1.カラムの性能をチェックする方法は?
カラムは消耗品ですので、丁寧に使っていても永年の使用で劣化してきます。定期的に性能のチェックをすることが重要です。
ODSカラムの場合のチェック方法を以下に紹介します。
- カラムの非保持容量(V0)を確認するために、カラムに保持されない化合物(ウラシル等の極性物質)を1つ用意します。
- カラムに保持される化合物(ベンゼン、ナフタレン、トルエン等)を最低2つ用意します。
- 以上の3成分をカラムに通し、理論段数(N)、保持係数(k)、分離係数(α)、アシンメトリー係数(As)を算出します(下記の式を参照)。

(診断方法)
- Nの値が小さくなってくると、充填材の汚染や,充填状態の悪化などカラムの劣化が考えられます。
- kの値が小さくなると、ODS基の脱落が考えられます。
- αの値が小さくなると、k値の低下のほか、充填材の汚染が考えられます。
- Asの値は1に近いほどよい状態と診断されますが、1からずれてくると、充填材の汚染、充填材の劣化などが考えられます。
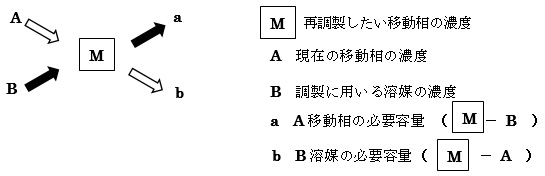
Q2.一度調製した移動相を用いて、異なる濃度の移動相を再調製したいのですが?
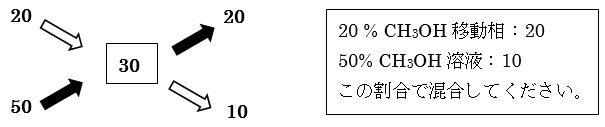
下図に数値を当てはめれば、簡単に調製できます。
(例1)20 % CH3OHの移動相から、50% CH3OHを用いて新たに30% CH3OHを調製したい。
(例2)35 % CH3OHの移動相から、水を用いて新たに25% CH3OHを調製したい。

Q3.逆相分配クロマトグラフィーを行う際、カラムは何を基準に選べばよいですか?
回答準備中です。
Q4.各メーカーから様々な種類の逆相分配カラムがでていますが、その中から自分にあったカラムを選ぶには?
回答準備中です。
Q5.逆相分配カラムで解離しやすい官能基を持つ試料を分離する際の注意点は?
カルボキシル基(-COOH)やアミノ基(-NH2)などの極性官能基の解離は、水素イオン濃度(pH)やイオン強度に大きく影響されます。したがって、逆相分配クロマトグラフィーの場合、移動相として極性の大きい水系溶媒を用いますので、この移動相のpHが解離しやすい官能基を持つ成分の分離に大きく影響するわけです。つまり、移動相のpHが低くなると、カルボキシル基のような酸性の官能基は解離が抑制され固定相への親和性が強くなるため保持係数(k)は大きくなり、アミノ基のような塩基性の官能基は解離が進み固定相への親和性が弱くなるためkは小さくなります。逆に移動相のpHが高くなると、酸性の官能基を持つ成分の解離は進み固定相への親和性が弱くなるためkは小さくなり、塩基性の官能基を持つ成分の解離は抑制され固定相への親和性が強くなるためkは大きくなります。
すなわち、逆相分配カラムで解離しやすい官能基を持つ成分を分離する場合は、官能基の解離を抑えて、成分が固定相に十分に保持される事が重要で、酸性の官能基を持つ成分を分離する場合は移動相のpHを下げ、塩基性の官能基を持つ成分の場合は移動相のpHを上げる方法(イオン抑制法)が取られます。
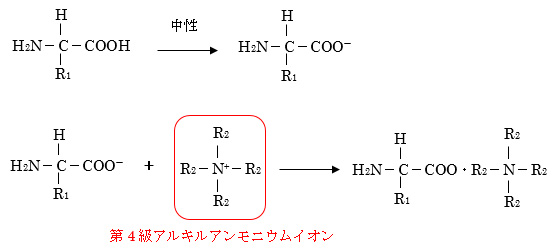
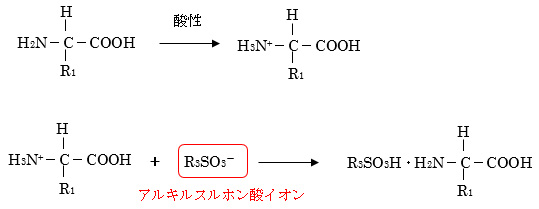
また、イオン抑制法以外に解離しやすい官能基を持つ成分を分離する方法として、イオン性の官能基とイオン対を形成する試薬を加えることで電荷を持たない、固定相に保持されやすい物質に変換し、分離を行うイオン対クロマトグラフィー(イオン対法)が開発されています。イオン対試薬としては、酸性の官能基を持つ成分に対しては、第4級アルキルアンモニウム塩、第3級アミンなどが、塩基性の官能基を持つ成分に対しては、アルキルスルホン酸塩、過塩素酸などが用いられます。
なお、イオン抑制法、イオン対法いずれの場合も、固定相担体の使用可能pH範囲内で行うように十分に注意してください。
Q6.イオンペアクロマトグラフィーについて教えてください。
Q7.イオンペア試薬の使い方について教えてください。
イオンペア(イオン対)クロマトグラフィーとは、水溶液中のイオン性物質に対し、それと逆の電荷を持つイオン(カウンターイオン)を添加してイオンペアを形成させ、全体の電荷を中和して低極性物質に変換することで、逆相分配カラムの固定相に保持されやすくする方法です。このとき使用するカウンターイオンをイオンペア(イオン対)試薬と呼びます。酸性物質に対しては、第4級アルキルアンモニウム塩、第3級アミンなどが、塩基性物質に対しては、アルキルスルホン酸塩、過塩素酸などが用いられます。
注意点としては、カラムが安定するのに少し時間が必要です。また、カラムからイオンペア試薬を洗い流す時、水を流す前にまず酸性緩衝液と有機溶媒の混合液を用いてください。
(例)酸性物質に対するイオンペア試薬(移動相が中性:pH7付近)
(例)塩基性物質に対するイオンペア試薬(移動相が酸性:pH2~3)
Q8.グラジエント分析法で、使用するカラムの長さは分離効率に影響しますか?
Q9.グラジエント分析法で、使用するカラムの内径は分離効率に影響しますか?
Q10.グラジエント分析法で、流速は分離効率に影響しますか?
イソクラティック分析法の場合、分離効率はカラムの長さに関係し、カラムが長いほど分離はよくなります。それに対して、グラジエント分析法では、グラジエント時間をカラムの長さに比例して変化させれば、分離効率はカラムの長さに影響を受けずほぼ同じです。例えば、カラムの長さが250 mmと150 mmの場合を考えてみると、250 mmのカラムで20分のグラジエント時間で得られた分離効率とほぼ同じものが、150 mmのカラムでは、250 mmのカラムの6割の12分で得られることになります。
カラム内径と分離効率の関係は、同一のグラジエント勾配条件、あるいは同一の流速では、カラム内径に比例して、それぞれ流速、あるいはグラジエント時間を調整すれば、同一の分離効率を得ることができます。つまり、同一のグラジエント勾配条件では、カラム内径を大きく(小さく)する場合は、流速を速く(遅く)します。また、同一の流速では、カラム内径を大きく(小さく)する場合は、グラジエント時間を長く(短く)します。
グラジエント時間と流速の関係は、同一のグラジエント勾配条件では、流速だけを変化させても同一の分離効率は得られませんので、流速に応じてグラジエント時間を調整する必要があります。つまり、流速を速く(遅く)する場合は、グラジエント時間を短く(長く)します。
Q11.カラム使用後の洗浄、保存方法を教えてください。
回答準備中です。
Q12.HPLCに使用する有機溶媒の取り扱い注意点は?
回答準備中です。
Q13.移動相の脱気は必要ですか?
混合溶媒作成を、LC装置内ではなく予め移動相ビン内で行うとき(イソクラティック)のお話です。
メタノールは、水と混ざると発熱し余計な溶存空気が気泡となって抜けやすく(脱気されやすく)なります。一方アセトニトリルでは、吸熱して冷えてしまうために、徐々に室温に戻るにつれて後から気泡が発生してしまいます。従って、より脱気(加温撹拌、膜脱気ユニット、Heパージなど)に配慮が必要です。
Q14.移動相に緩衝液を使用する際の条件と注意点は?
回答準備中です。
Q15.よいカラムとはどんなカラムですか?
回答準備中です。
Q16.生体試料中に夾雑している高分子物質(たんぱく質、多糖類など)を除く方法は?
回答準備中です。
Q17.分離ピークのテーリング、リーディング、ブロード化の原因は?
ピーク形状が異常になる原因はいろいろ考えられます。
【溶離液】
溶離液の組成(溶媒組成、pH、緩衝液濃度など)がピークの形状に影響を与える場合があります。塩基性化合物を逆相分配カラムで分析する場合、中性の溶離液ではテーリングを起こしていたものが、溶離液pHを酸性に下げることで改善されることがあります。例えば、移動相に酸(トリフルオロ酢酸や酢酸など)を0.1-1%程度添加します。これは、中性付近で起きている固定相の残存シラノール基の解離が、酸性にすることで抑えられ、化合物との相互作用が低減されたことによると考えられます。酸性化合物を逆相分配カラムで分析する場合は、対象化合物のpKa値より低いpHの溶離液を用いれば、テーリングの解消が期待できます。また、ODSカラムの場合、オクタデシル化後に残ったシラノール基をより小さな分子のシリル化剤(例えば、トリメチルシリル化剤)で処理(エンドキャッピング)したカラムを使えば、残存シラノール基との吸着性が強いサンプルのピーク形状改善が期待できます。塩基性化合物の分離には、溶離液へのアミン類などの抗テーリング剤の添加も有効です。
固定相に含まれる微量の金属とのキレートが原因でテーリングを起こすことがあります。そのような金属キレート性成分の分離には、移動相に5mmol/L程度のEDTA・2Na(エチレンジアミン四酢酸二水素二ナトリウム)を加えると分離が改善することがあります。
また調製後長時間経過した溶離液には異物が混入したり、微生物が繁殖したりすることがありますので、定期的に溶離液を新しく調製し直しましょう。
【ポンプ】
ピークがブロードになったり溶出時間が安定しない場合、ポンプのトラブルで溶離液の流速が設定どおりでなかったり、配管などから溶離液がリークしていることが考えられます。また、圧力が高い場合はカラムが原因である場合がありますので、後で述べる方法で対処します。
【試料】
試料の分解が原因として考えられます。また、溶離液と比べて溶出力やpH の差が大きい試料溶媒を大量に注入すると、リーディングやショルダー、ピーク割れを起こす場合がありますので、試料は溶離液に溶かしてください。溶けない場合は、溶解する溶媒に溶かした後、溶離液で希釈してください。また、注入量もなるべく少なく、半分以下程度に減らしてください。
【カラム】
カラムの劣化が原因で、ピーク形状の異常、ゴーストピークの出現、圧力上昇が起こります。ガードカラムを装着している場合は、これを外して回復すればガードカラムの劣化が原因ですのでガードカラムを交換します。回復しない場合は分析カラムの劣化が原因と考えられます。分析カラムの劣化には、①カラム入口側に発生する隙間、②固定相の不均一な充てん状態による溶離液の不均一な流れ、③固定相充てん剤表面の修飾基の分解・剥落、④主に入口付近の不純物や高濃度試料による汚染があります。①~③が原因の場合はそのカラムは通常回復しませんので新品のカラムに交換します。④の場合は各カラムの取扱説明書などを参考にカラムを洗浄します。カラムの洗浄を行なっても回復しない場合は新品のカラムに交換します。
カラム温度が設定からずれるとピークの溶出時間が変わる場合があります。またカラムや溶離液の交換時などコンディショニングが不十分な場合や、カラムと検出器を接続する配管が長すぎたり内径が大きすぎたりするとピークがブロードになる場合があります。
ゴーストピークの出現の原因もいくつか考えられます。
溶離液中の水に含まれる不純物ピークが出現している可能性があります。カラム平衡時にカラムに保持された不純物が、グラジエント分析中に移動相の溶出力が上がっていくと溶出され、サンプルと同様にピークとして現れることがあります。
目的成分以上に固定相への親和性が強い共雑成分が含まれている場合、前回打ったサンプル中の共雑成分が溶出していない可能性があり、これがゴーストピークとして出現することがあります。逆相分配カラムの場合では、分析に用いた溶離液の有機溶媒濃度以上の濃度の有機溶媒でカラムを十分に洗浄してください。
装置の汚れによりピークが出現している可能性があります。 特に、オートインジェクターのニードルやマニュアルインジェクターの場合のシリンジは汚れやすいので、定期的に汚れを落とすような溶媒で超音波洗浄することをお勧めします。インジェクター内が汚れている場合は、インジェクション毎にピーク面積の減少が見られます。その場合も、インジェクターの汚れを溶解するような溶媒をシリンジを用いて注入して洗浄してください。
サンプル調製用溶媒と溶離液の組成が異なる場合、サンプル溶媒がピークとして現れることがあります。この場合は、サンプルを溶離液と同じ組成の溶媒で溶解してください。溶けない場合は、溶解する溶媒でいったん溶かした後、溶離液で希釈してください。
Q18.高極性物質をHPLCにより分析したいのですが、どのような方法がありますか?
近年、HILIC(Hydrophilic Interaction Chromatographyの略称)という分離モードが注目されるようになってきています。日本語では「親水性相互作用クロマトグラフィー」と呼ばれています。HILIC は順相クロマトグラフィーの一種ですが、ヘキサンなどの水と混和しない溶媒は溶離液に使用しません。HILICモードは、極性基を有する固定相と逆相系で使用するCH3CN/CH3OH/H2O系移動相の組み合わせにより、親水性化合物を固定相に保持させ分離する方法です。HILICモードでは親水性の固定相表面に水和相が形成され、試料成分の分配は移動相と水和相の間で起こります。したがって、水和相に親和性の高い高極性化合物が保持される一方、低極性化合物はほとんど保持されず早く溶出します。また、移動相の有機溶媒濃度を増加させ溶離液の極性を下げることで、極性化合物の保持が大きくなります。例えば、溶媒の溶出力の強さは、水(H2O) > メタノール(MeOH) > エタノール(EtOH) > イソプロパノール(IPA) > アセトニトリル(ACN) > アセトンとなりますので、極性化合物の保持力の強さは、ACN:H2O=90:10 < ACN:H2O:MeOH=90:5:5 < ACN:H2O:EtOH=90:5:5 < ACN:H2O:IPA=90:5:5となります。
HILICの利点として、①逆相カラムでは保持されない高極性化合物の保持が可能で、低極性のマトリックス成分の妨害が少ない。②移動相にアセトニトリルなどの揮発性の有機溶媒を高濃度に使うので、MS検出器に接続した場合に、移動相の水含有率が高い逆相系と比較してイオン化効率が上昇し感度の向上が期待できる。つまり、HILICはLC-MSに適したモードと言える。などが上げられます。
Q19.逆相カラムには多くの種類が市販されていますが、違いを教えてください。
逆相カラムでは、ODSカラムが最も一般的ですが、C18(オクタデシル基)は鎖の長さが長く疎水性が強いので、サンプル中の疎水性の強い成分が長く留まりすぎ、測定時間が長くかかりすぎることがあります。このため、鎖の長さのもっと短いC8(オクチル基)、C4(ブチル基)、C3(トリメチル基)などが結合されたカラムが市販されており、成分の疎水性の違いにより使い分けます。また、このほかにフェニル基、シアノプロピル基などが結合されたシリカゲルを充てんしたカラムもあり、成分の保持力は弱いですが、アルキル基の結合性とは異なる特異的な選択性が期待できます。
Q20.HPLCでアミノ酸を分析したいのですが、どのような方法がありますか?
アミノ酸を検出するには、ベンゼン環を持つアミノ酸は250~280nmでの検出も可能ですが、一般にはカルボキシル基の200~210nmでの吸収を利用するしかありませんので、そのままの形では高感度に選択性良く分析するのは困難です。
そこで、アミノ酸のアミノ基に対して選択的に反応する誘導体化試薬を用いた誘導体化法が古くから使われており、プレカラム誘導体化法とポストカラム誘導体化法に分けられます。
【プレカラム誘導体化法】
試料を注入する前にアミノ酸を誘導体化し、その誘導体化アミノ酸をHPLCで分離・検出する方法です。代表的なプレカラム誘導体化試薬として、フェニルイソチオシアネート(PITC)(UV検出)、o-フタルアルデヒド(蛍光検出)、フルオレサミン(蛍光検出)、ダンシルクロライド(蛍光検出)、ベンゾフラザン試薬(光学活性アミノ基の蛍光検出)などが挙げられます。この方法の利点、欠点は以下の通りです。
利点
1) 装置構成がシンプルで、反応系を小さく設定できるので試薬消費量が少なくてすむ。
2) 事前に誘導体化試薬と十分に反応させることで、高感度化が可能となる。
3) 検出器の種類に応じて、誘導体化試薬が選べる。
4) 分離モードの主流である逆相系が適用できるので、迅速分析が可能となる。
欠点
1) 誘導体化効率が、試料中の共雑物や溶媒などの影響を受けやすい。
2) 誘導体が不安定な場合は、定量結果に影響が出る。
【ポストカラム誘導体化法】
カラムでアミノ酸を分離した後、誘導体化試薬と反応させて検出する方法です。
最もよく用いられる分離モードは、陽イオン交換です。 強酸性アミノ酸ほど早く溶出し、塩基性アミノ酸ほど遅く溶出します。また、共雑する恐れのあるアミン類は、ほとんどの場合アミノ酸よりは塩基性が強く、したがって陽イオン交換においてはアミノ酸類よりも遅く溶出しますので、アミン類がアミノ酸の定量を妨害する心配はありません。親水性の高いアミノ酸は、逆相法では十分な保持や選択性が得られない可能性がありますが、 陽イオン交換法を使うことによって、アミノ酸同士やアミン類との分離が容易に行えます。
代表的なポストカラム誘導体化試薬として、ニンヒドリン(VIS検出)とo-フタルアルデヒド(蛍光検出)が挙げられます。この方法の利点、欠点は以下の通りです。
利点
1) 反応が自動化できるため、定量性、再現性に優れる。
2) 反応前に試料中の共雑成分がカラムで分離されるため、誘導体化効率反応効率が上がる。
欠点
1) 高感度化が難しい。
2) 試薬消費量が多くなる。
3) 未反応試薬も検出器に流れ込むので、それが検出されてはならないという制限があり、使用できる試薬の種類が限られる。
4) 分離モードの主流である逆相系が適用できないので、分析に時間がかかる。
Q21.HPLCで陰イオン界面活性剤を分析したいのですが、どのような方法がありますか?
回答準備中です。
Q22.HPLCで非イオン界面活性剤を分析したいのですが、どのような方法がありますか?
回答準備中です。
Q23.HPLCで雨水中の無機イオンを分析したいのですが、どのような方法がありますか?
近年、大気汚染の進行に伴い、その主な原因物質である大気中のSOx,NOxが雨水中でH2SO4(硫酸)、HNO3(硝酸)などに変化することで酸性雨となり、生態系への影響が問題となっています。これらの無機イオンの分析には、高感度、高精度分析が可能なイオンクロマトグラフィー(IC)が広く用いられていますが、逆相分配カラムを用いた逆相イオンペアクロマトグラフィーも使用されています。特に、ICは電気伝導度検出器(CD)との組み合わせにより、高感度分析が可能となっており、雨水、河川水、上水、地下水などの環境水中の陰イオン、陽イオン分析に威力を発揮しています。
ICには、分離カラムとCDの間にサプレッサーを配置し、検出段階で溶離液を電気伝導度の低い溶液に変換するサプレッサー方式と、サプレッサーを配置せずに電気伝導度の低い溶離液に用いるノンサプレッサー方式の2つがある。
Q24.HPLCで糖類を分析したいのですが、どのような方法がありますか?
糖類には、単糖、オリゴ糖、多糖、中性糖、酸性糖、アミノ糖、糖アルコール、糖ペプチドさらにそれらの異性体など様々な形態が存在し、分子量の幅も大きく、しかも発色団を持たないため、HPLCによる一斉高感度分析が難しいとされています。したがってHPLC分析にあたっては目的に応じて適切な分離法や検出法を選択する必要があります。
【分離法】
主な分離モードには、サイズ排除、配位子交換、分配(順相)、陰イオン交換、HILICなどが挙げられます。それぞれの特徴を説明します。
サイズ排除:糖類を分子量により分離したい場合に用います。基本的には分子サイズによる分離ですので、高分子量の多糖類には適していますが、同一分子量の単糖は分離できません。
配位子交換:親水性ポリマーにNa+、Ca2+、Pb2+などの金属対イオンを結合させた充てん剤を用い、主として糖類の水酸基と金属対イオンとの錯形成に基づいて分離する方法ですが、分離のベースは、排除限界分子量約1000のサイズ排除法ですので、分子量の大きな糖から順番に溶出されます。主に2糖類以下の糖類の分離に適しており、金属対イオンの種類と糖類のもつ水酸基の位置や数により同じ分子量の単糖類でも錯形成能に差が出てきますので、お互いを分離することが可能になります。移動相に使用するのは水だけですので、移動相による分離のコントロールはできません。
分配(順相):一般にシリカポリマー坦体にアミノプロピル基を結合させた充てん剤が用いられます。単糖類からオリゴ糖までの分離に適しており、特に、オリゴ糖構成糖の違いを識別することができますので、2糖類の相互分離も可能です。移動相にはアセトニトリル/水系が用いられ、水の比率が増えるほど、また、糖類の分子量が小さいものほど早く溶出します。なお、固定相のアミノ基は糖類のアルデヒド基と反応してシッフ塩基を作りますので、カラムへの吸着力が強くなり、特に五単糖(アラビノース、リボース等)ではピークが大きくテーリングすることがあります。これは、移動相に塩を添加することにより抑制することができます。また、単糖同士の相互分離には限界があります。
陰イオン交換:中性糖のpKaは12程度ですから、強塩基性条件下では糖のアルコール性水酸基の一部あるいはすべてが解離してイオン性分子として存在しますので、陰イオン交換樹脂に保持させることができます。この場合、移動相としては強塩基性水溶液(例えば、0.1M程度の水酸化ナトリウム溶液)が用いられます。一般に、単糖類からオリゴ糖の順に溶出します。水酸化ナトリウム濃度を変化させるグラジエント法と組み合わせますと、一斉分離が可能となります。
HILIC:近年、生体由来の親水性化合物の分離にHILICが見直されてきており、オリゴ糖や糖ペプチドにも応用が広がっています。理由としては、逆相分配系の移動相溶離液とほぼ同じ組成条件で、逆相系では保持されにくい親水性化合物の分離が可能となったことが挙げられます。さらに、高選択性、高感度な検出器として普及してきた質量分析計(MS)にとって、揮発性の有機溶媒の比率が高く、高濃度の塩を含まない溶離液は、MSにおけるイオン生成の上で有利であることも理由の一つです。充填剤には、従来からのシリカ、アミン、アミド系に加えて、ポリアミン、ポリアクリル酸、ポリビニルなどが開発されています。
【検出法】
主な検出法には、紫外可視検出、示差屈折率検出、蛍光検出、電気化学検出などが挙げられます。それぞれの特徴を説明します。
紫外可視検出:糖類の紫外吸収は190~195nm付近の低波長域に限られますので、直接紫外検出を行うのは実用上困難です。誘導体化して紫外可視検出する方法もほとんど使用されておりません。
示差屈折率(RI)検出:試料成分と移動相溶離液の間に屈折率の差があれば何でも検出できる汎用検出法です。RIレスポンスが成分濃度に比例し、検出感度も従来に比べて向上してきたことから、糖類の分析には広く用いられています。しかし、汎用検出法ですので検出の選択性は低く、また、RIに影響を及ぼすグラジエント溶出法が使えませんので、多成分一斉分離には有効ではありません。
蛍光検出:蛍光検出法は、高感度で高選択性の優れた検出法ですが、糖類は蛍光性ではありませんので、蛍光誘導体化が必要です。蛍光誘導体化法には、プレカラム方式とポストカラム方式があり、それぞれの特徴を説明します。
・プレカラム方式
誘導体化試薬としては、糖の還元末端と反応性の高い2-アミノピリジンやローダミン系色素が用いられています。誘導体化効率が高く、高感度検出が可能ですが、誘導体化処理にある程度の時間と手間が必要です。
・ポストカラム方式
誘導体化反応の自動化という面で優れていますが、誘導体化反応が迅速で、選択性が高く、高感度であることが要求されます。そういう観点から、誘導体化試薬としては、2-シアノアセタミド、エタノールアミン、アルギニン/ほう酸などが用いられていますが、プレカラム方式に比べて誘導体化効率が悪く、また、反応時間、温度、試薬の影響などの点で制約を受けやすいので、感度も低くなります。
電気化学検出:検出器セル内の電極表面で起こる酸化還元反応で生じる電位差(電流値)を測定する方法です。糖類の検出には、主に金電極を用いますが、特に、金電極にパルス電位を繰り返し印加する方法は、糖の高感度検出を可能にします。ただし、反応液を強アルカリに保つ必要があるため、移動相条件によっては、カラム溶出液に高濃度の水酸化ナトリウムを添加することが必要になります。また、酸化還元反応を起こす夾雑成分の妨害が問題になることがあります。
Q25.逆相HPLCの溶離液によく使われるメタノールとアセトニトリルの用途の違いは何ですか?
アセトニトリルは、粘度が低く、UV吸収が低いため、逆相HPLCにおいて移動相としてよく用いられます。
【保持時間】
アセトニトリルはメタノールに比べ、極性が低いので、メタノールへ変更すると、保持時間が長くなります。
アセトニトリルとメタノールを各々同じ比率で水に混ぜ合わせた場合、一般にアセトニトリル系の方が溶出力が強くなります。特に混合比率が低い時には、同じ保持時間を得るのにアセトニトリル比がメタノールの半分以下で済むことがあります。
【カラム圧力】
カラム圧力はメタノールの方が高くなります。特に有機溶媒比率が60%のとき、メタノールはアセトニトリルの約2倍のカラム圧力になります。分析温度を上げると、カラム圧力を抑えることができ、また、保持時間が短くなりピーク形状も良くなります。しかし分析温度を上げると、カラム劣化を促進させるので、より高耐久性のカラムが必要とされます。
メタノールは水との混合で圧力が高くなりますが、アセトニトリルはそれ程でもありません。アセトニトリル系の方が、同じ流速ではカラムに余計な圧力がかかりません。
【UV吸収】
メタノールは低波長付近においてアセトニトリルより大きな吸収を持つので210~230 nm 付近でS/N比が高くなり、微量分析やグラジエント分析には適しません。但しHPLC用のメタノールを用いることで、多少はS/N比の改善ができます。
【溶出順序の違い】
逆相HPLCでは、移動相の有機溶媒比率が高くなれば保持時間は短くなります。物質によっては、アセトニトリルとメタノールでは溶出順序が逆転する場合があります(例:安息香酸とアニリン)。これは、アセトニトリルが非プロトン性、メタノールがプロトン性によるものであるといわれており、疎水性相互作用と同時に,プロトン親和性が影響することがあります。例えば安息香酸の-COOH基のようにプロトン供与性の官能基をもつ化合物では、-COOH基の水素とアセトニトリルのCN基の窒素の孤立電子対が引き合うため、水/アセトニトリル系の方が水/メタノール系より溶出が早められることがあります。一方、アニリンのような芳香環に-NH2基をもつ化合物については、プロトン性メタノールの水素と-NH2基の窒素の孤立電子対が引き合うために、水/メタノール系の方が水/アセトニトリル系より溶出が早められることがあります。
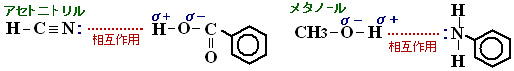
【ピーク形状】
塩基性医薬品など吸着しやすい物質の分析では、移動相の有機溶媒をアセトニトリルからメタノールに変更すると、ピークのテーリングが小さくなります。これは移動相の有機溶媒が残存シラノール基と分析対象物質との相互作用に影響を与えていると考えられます。
アセトニトリル系ではテーリングが大きく、メタノール系では抑制される、という場合があります。サリチル酸のような(オルト位にカルボキシル基やメトキシ基を持つフェノール)化合物などでこの傾向が認められます。これはシリカ表面と目的成分との(吸着)相互作用に対する移動相の関わり方が、有機溶媒分子の化学的性質によって異なるためと考えられます。また、ポリマー系逆相カラムでは、アセトニトリル系でテーリング傾向が目立ちにくい場合が多くあります。
【試料の溶解性】
メタノールは、酸・塩基性化合物などのイオン性試料に対してアセトニトリルより高い溶解度を示します。溶解性の高い溶媒で調整した試料では、注入量を減らしたり、移動相と試料との有機溶媒濃度を合わせることが容易です。
Q26.カラムを長持ちさせる方法は?
回答準備中です。
Q27.カラムの圧力が上昇してしまいました。その原因と対処法は?
回答準備中です。
Q28.逆相HPLCで使用する溶離液中の有機溶媒の割合と保持時間との関係は?
回答準備中です。
Q29.順相HPLCで使用する溶離液中の有機溶媒の割合と保持時間との関係は?
回答準備中です。
Q30.分析開始前、溶離液をどの程度流せばカラムが安定しますか?
回答準備中です。
Q31.カラムを初めて使用する場合の取り扱い方、カラム使用後の洗浄、保存方法を教えてください。
HPLC を行う際、市販のパックドカラムを使用することがほとんどだと思います。この場合、出荷時にカラム内に充填されている溶媒から実際使用する溶離液に置換する必要があります。また使用後、使用した溶離液のまま長期間カラムを置いておくとカラムの劣化につながることがあるため、カラム内を適当な溶媒に置換する必要があります。
【出荷時充填溶媒から使用溶離液への置換】
(1)まず出荷時のカラム内充填溶媒の種類を確認して下さい。溶離液と互いに混和しない溶媒の場合、両方に混和する溶媒で置換後、溶離液に置換して下さい。溶離液に緩衝溶液を用いる場合、緩衝溶液を構成する塩類によってはカラム内の有機溶媒によって析出しカラム劣化の原因となる場合がありますので、あらかじめ水などの塩が溶解する溶媒に置換するなどの配慮が重要です。
(2)カラム内の使用溶離液への置換はカラム出口側をオープンにして行い、カラムの接続は、HPLC 装置内も溶離液で十分置換してから行って下さい。
【カラム使用後の洗浄、保存(短期)】
(1)実際使用した分離条件では、分析対象成分以外の共雑物がカラム内に留まっている可能性がありますので、溶出力のある溶媒で十分に洗浄してください。数日間の短期であれば、カラムは接続したままで構いません。
(2)緩衝液を使用した場合は、水などの塩類が溶解する溶媒を流し塩類をカラム内から取り除いて下さい。
【カラムの保存(長期)】
出荷時充填溶媒に置換した後ラインから外し必ず密栓をしてカラム内を乾燥させないように保存してください。しかし長期保存する場合でも、1,2ヶ月に一度はカラム内を通液して下さい。
※これからも改版を重ねてまいりますので、どうぞよろしくお願い致します。
Ver1.0
![]()
